 The CRYSTALS v14.6236 installer is now available.
The CRYSTALS v14.6236 installer is now available.
This update fixes a number of bugs and eliminates a few mysterious crashes. Please report problems. The minor component or the version number now refers to a specific snapshot of the source code enabling us to better identify and fix bugs.
Key changes between v14.5841 and 14.6236
Ensure copied text is flushed to clipboard on CRYSTALS exit (otherwise it gets lost).
Upgrade xinvert script to accommodate the 11 enantiomorphic space groups and the 7 which require origin shifts.
Diffin: Set default for Z in case no formula found.
Diffin: Extend filename length to 512 to help when running in folders with long paths
Catch crash on matrix inversion of singular matrices.
Xcif: Disable ‘Include HKLI’ and ‘Include squeeze’ if include files not present in folder.
Xafour: Change limiting density question to a dialog to avoid new user confusion.
Close xpublish window after CIF dialog is dismissed.
Add CHANGE RESIDUE to pop-up menus
Speed up plots of internal vs external sigmas
Fix style and centre of rotation of zoomed objects. Add more ‘zoom’ and ‘unzoom’ options to menus
Improve popup text on model toolbar buttons
Add SWAP directive to #EDIT. This swaps the atomic and thermal parameters of pairs of atoms.
Insert residues numbers of generated hydrogen atoms
Sanity check of parameter values when loading from disk. Replace NaN with 0.0 or 1.0 as appropriate.
Show scale factor for electron densities, fix NANs in(Fo^20Fc^2) pattersons.
Cameron: Keep outline of small atoms at smaller scales.
Added header to design matrix ASCII output
Tabbed plot of Internal vs External variances
Insert appropriate occupancy and part # for H’s adjacent to split groups.
Spot clashing H-serial numbers on the fly and correct.
Increase allocation for lexical list processing (was falling over at 500 CONTINUATION lines, should now stretch to 2000).
Extend scattering factor look-up table out to Rho = 3.0
Enable unusual anomalous scattering factors to be input directly from the Cell/SG tab.
Add code to List 12 processing to catch singularities in rigid body code. Least useful rotational parameters are removed from the least-squares.
Enable LIST 2 to be output in full (#PUNCH 2) or just as the symbol (#PUNCH 2 B). This latter enables CRYSTALS to create an appropriate LIST 14 on re-input, otherwise the user must create it.
Changed handling of reflection sigmas. #LIST 6 now has an extra item, L30UP, which controls whether LIST 30 is updated as reflections are read in.
xregulh left SCPQ open, which meant that xwrite5 failed to delete it, resulting in the H optimisation list of refinable atoms being left in there and used to setup L12 if, and only if, there were no OH or NH atoms to be refined. FIXED.
Context menu entry to swap two atom labels.
Read SHELX T .res files (ignore peak height – last column in atom-coordinate list)
Added EXPORT button to FO vs FC graph (csv format).
Extended support for 6-digit H atom serial numbers to xwrite5.scp (not supported everywhere, but here is quite critical).
Improve treatment of A&B parts. Phase shift is included in A&B as they are stored for compatibility with PLATON SQUEEZE, then removed as they are used and the Phase shift applied separately.
Provide clear warning when link for PLATON fails because of a missing LIST
Fix reported cell volume e.s.d for tetragonal and cubic systems (includes correlation of length parameters).
NEW #PUNCH 6 I punches all stored keys.
Use bond list for auto H addition. Use part numbers to generate multiple H conformations on nearby atoms.
Manual ADD H (right-click Add hydrogens) now puts the new H’s into a sensible PART based on connected atom parts. Quite useful.
Allow automatic ‘ride’ of multiple part H’s on same carbon atom (previous version repeated the pivot atom in the constraints list).
Keeping CheckCIF happy: Suppress ESD output on H-bond angle (D-H–A) if the H is riding or fixed.
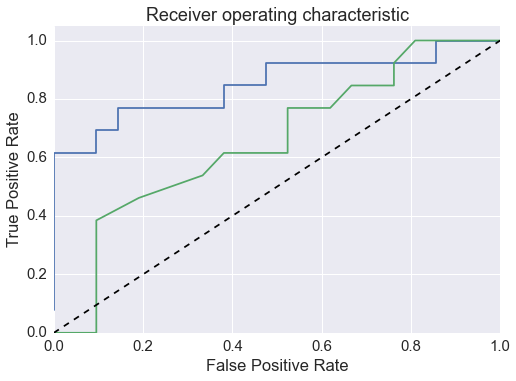
Add KEYS to reflection statistic plots. Add +/- 10% bands to npp.
 The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.
The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.