The CRYSTALS 15.0.1 (v7809) installer for Windows 7/10 is now available. This is a quick patch release following the release of v7795.
Please report problems. The version number refers to a specific snapshot of the source code enabling us to better identify and fix bugs.
Key changes between 15.7795 and 15.7809
This is a small patch release which fixes a couple of broken URLs, adds some new icons, new shortcut keys, quick access to import and export via toolbar.
Key changes between 14.6999 and 15.7795
This release of CRYSTALS has changed the way the software is installed, so that you don’t need administrator permission to install or run it.
If you have an old version of CRYSTALS installed, and you follow the prompt to uninstall it, it will require administrator permissions. If you do not uninstall the old version there may be duplicated information in different parts of the registry, but it should not prevent use of the software.
The new install structure brings CRYSTALS closer to best practice for installing applications on Windows, and as a result you can no longer find the application and examples in c:\wincrys\
While this will be distressing for users from the days of the command line, it shouldn’t affect point-and-click users very much. Application and supporting files can be found in
%LOCALAPPDATA%\Programs\crystals
and demo files are in
%APPDATA%\crystals\demo
If you need to access demo files or edit CRYSTALS scripts, you can paste the locations above into a Windows Explorer, or find shortcuts to both of these locations in the Windows start menu in the Crystals group.
Other changes since v14.6999 (July 2018):
- Added ‘cycle residue’ button to toolbar. Eases working with large structures. See: https://www.youtube.com/watch?v=sN7w-THNmTo
- Added ‘cycle parts’ button to help visualise disorder parts: https://youtu.be/OoRm9J7hfjE?t=268
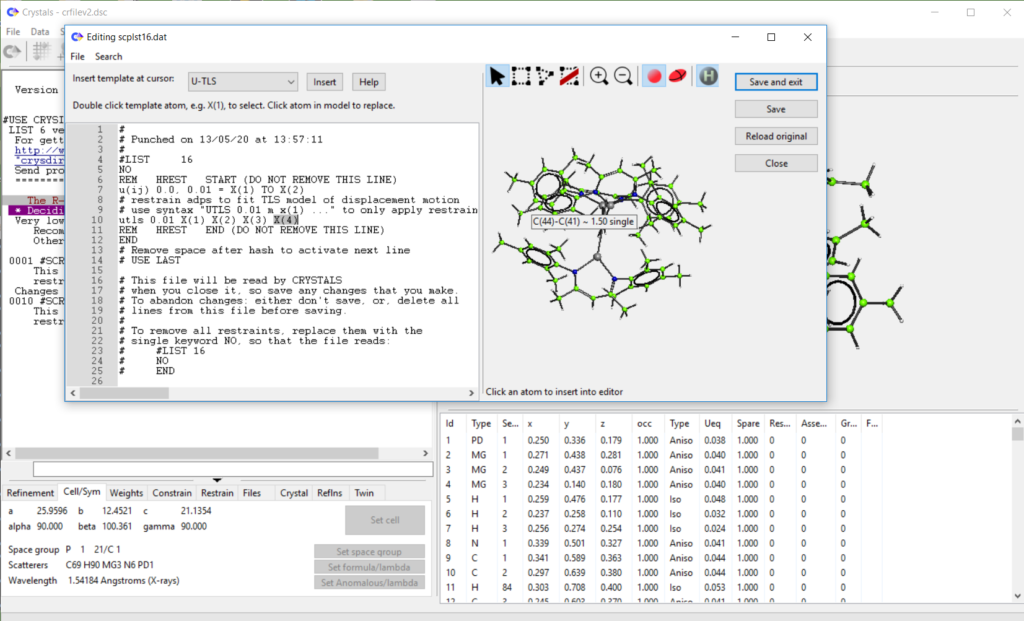
- Added ‘restrain templates’ to list 16 editor. Work in progress, see demo here: https://www.youtube.com/watch?v=R3SHqKv9pCg
- Improved refinement mode override for individual atoms. Video here: https://www.youtube.com/watch?v=sCTV4HN8RDk
- Catch instabilities during matrix inversion, allow user to choose precision or speed in GUI
- Improved undo structure dialog. Catch cases with lists marked as ‘in error’. Structure is shown automatically when selecting.
- Reflections with a weight of zero are excluded from the total count.
- Output files (listing, punch, mon) and many intermediate data files are now stored in subfolders of your working directory to keep it tidier.
- Additional colours added to colour tables for atom types.
- Removed dependency on CRYSDIR environment variable when launched from command line. Will override if set, but otherwise .exe location is used.
- Sort deuterium with H in CIF formula.
- Peak height slider added to find H dialog, use it to hide low density peaks. Improved usage with neutron H maps.
- Slant Fourier dialog now computes default size limits which encapsulate the selected atoms.
- Fix cell su issue when importing data from .ins files (and other bugfixes for edge cases in shelx2cry)
- Fix sorting of data in Mogul dialog window (regression)
- Fix reading of squeeze data from PLATON (regression)
- Fix Cameron (regression)
- Changed behaviour: if you click a link to a file in the working folder in CRYSTALS text window (e.g. publish.cif) it opens in Windows Explorer instead of the default application.
- Upgraded MCE.
- Text output simplified, especially SFLS.
- Added Find and Replace dialogs to internal text file editor (e.g. for editing L12 and L16).
- Updated icons.
- Get WinGX location from the registry rather than environment variable.
- Historically Fourier map units are scaled by 10.0 to give 10 x e-/A^3 (e.g. in MCE or MapView). Command line default remains same for compatibility, but the GUI difference Fourier dialog now uses a scale factor of 1.0
- Added logging of file closures in .log file – easier to track which script or input file commands were from.
- Shelx2cry will now automatically import hkl if given a path to a different folder.
- Some renaming of CIF data items for filtered sets of data for compatibility with CheckCIF. All output is still present, comments in CIF explain the reasoning.
- Added electron radiation type. Scattering factors (eight-term approximation) for electrons included L.-M. Peng (1999) Micron 30, 625-648.
- Remember preference (per-structure) for refining occupancy of PARTS when using the Guide (saves unchecking box for every refinement setup).
- Default CRYSTALS merge updated to ignore large outliers. Will effect some noisy datasets if they are re-imported.
- Fixed Invert structure option in Structure menu.
- Fixed ‘change to uequiv’ popup menu
- Fixed ‘set Uiso’ popup menu item
- Many small bug fixes and improvements. Full commit history and changes at https://github.com/ChemCryst/crystals/

 The
The 