 Gwenno is investigating methods for determination of flexible organic molecular structures using the atomic pair distribution function.
Gwenno is investigating methods for determination of flexible organic molecular structures using the atomic pair distribution function.
Around labs, Gwenno manages to fit in singing, acting, dancing and painting, and enjoys skiing.
Gwenno is investigating methods for determination of flexible organic molecular structures using the atomic pair distribution function.
Around labs, Gwenno manages to fit in singing, acting, dancing and painting, and enjoys skiing.
 Oli Bar is investigating geometric parameters of metal organic frameworks using data analysis tools and machine learning.
Oli Bar is investigating geometric parameters of metal organic frameworks using data analysis tools and machine learning.
To unwind after a hard day of Python algorithms he plays tennis and badminton, and enjoys swimming. He prefers the non-standard setting of space group 14, P21/n, and at the time of writing he is undecided on his favourite exchange correlation functional.
 Kiaora’s research project is to determine the structures of molecules which are liquids at room temperature or otherwise hard-to-crystallize using crystalline host frameworks and/or low temperature in-situ crystallization techniques.
Kiaora’s research project is to determine the structures of molecules which are liquids at room temperature or otherwise hard-to-crystallize using crystalline host frameworks and/or low temperature in-situ crystallization techniques.
While away from the lab Kiaora indulges in the incompatible sports of baking and swimming.
Her favourite space group is P21/c and she prefers the hydrogen bond over other types of intermolecular interaction.
 James is carrying out a research project using molecular dynamics to improve our models of disorder in crystal structures.
James is carrying out a research project using molecular dynamics to improve our models of disorder in crystal structures.
James’ favourite undergraduate course was Prof Ritchie’s Quantum Mechanics stuff, and his preferred exchange correlation functional is the classic PBE.
 A big welcome to Chem Cryst to our new cohort of Part II students for 2017/18: Kiaora Tolmie, Lewis Morgan, Gwenno Jones, Han Xu, James Bird and Oli Bar! And welcome back to one of last year’s Part II students, George Sackman, as he starts his D.Phil. studies.
A big welcome to Chem Cryst to our new cohort of Part II students for 2017/18: Kiaora Tolmie, Lewis Morgan, Gwenno Jones, Han Xu, James Bird and Oli Bar! And welcome back to one of last year’s Part II students, George Sackman, as he starts his D.Phil. studies.
 The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.
The triennial congress of the International Union of Crystallography was held in Hyderabad over 8 days in August 2017.
During the meeting Richard Cooper presented recent work with Jerome Wicker:
Optimizing co-crystal  screens using a data-driven machine learning method
We also took an opportunity to present recent developments in the CRYSTALS software at the IUCr parallel programme Software Fayre:
Advanced restraints in CRYSTALS
Prior to the IUCr meeting, Richard Cooper was a tutor at the Crystallographic Computing School at IISc in Bangalore, organised by the IUCr Commission on Crystallographic Computing, and gave a lecture:
Recent advances in small molecule refinement

Photograph by @LouiseDawe
CrystEngComm, 2017, 19, 5336 – 5340 [ doi:10.1039/C7CE00587C ]
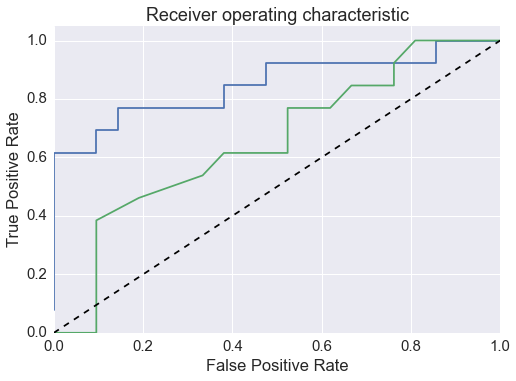 A data-driven approach to predicting co-crystal formation reduces the number of experiments required to successfully produce new co-crystals. A machine learning algorithm trained on an in-house set of co-crystallization experiments results in a 2.6-fold enrichment of successful co-crystal formation in a ranked list of co-formers, using an unseen set of paracetamol test experiments.
A data-driven approach to predicting co-crystal formation reduces the number of experiments required to successfully produce new co-crystals. A machine learning algorithm trained on an in-house set of co-crystallization experiments results in a 2.6-fold enrichment of successful co-crystal formation in a ranked list of co-formers, using an unseen set of paracetamol test experiments.
CrystEngComm, 2017,19, 3737-3745 [ doi:10.1039/C7CE00738H ]
 We present here the crystallisation outcomes for 319 publicly available compounds in up to 18 different solvents spread over 5710 individual single solvent evaporation trials. The recorded data is part of a much larger, corresponding in-house database and includes both positive as well as negative crystallisation outcomes. Such data can be used for statistical analyses of solvent performances, machine learning approaches or investigation of the crystallisation behaviour in structurally similar compound classes.
We present here the crystallisation outcomes for 319 publicly available compounds in up to 18 different solvents spread over 5710 individual single solvent evaporation trials. The recorded data is part of a much larger, corresponding in-house database and includes both positive as well as negative crystallisation outcomes. Such data can be used for statistical analyses of solvent performances, machine learning approaches or investigation of the crystallisation behaviour in structurally similar compound classes.
The presented data suggests that crystallisation behaviour in different solvents is not correlated with chemical similarity among clusters of highly similar compounds. Further, our machine learning models can be used to guide the solvent choice when crystallising a compound. In a retrospective evaluation, these models proved potent to reduce the workload to a third of our initial protocol, while still guaranteeing crystallisation success rates greater than 92%.
Publisher’s copy
 Congratulations to Part II students Laura Fenwick and James Walker who both won poster prizes awarded by the Chemical Crystallography Group of the British Crystallographic Association at the Annual Spring Meeting in Lancaster.
Congratulations to Part II students Laura Fenwick and James Walker who both won poster prizes awarded by the Chemical Crystallography Group of the British Crystallographic Association at the Annual Spring Meeting in Lancaster.
Laura’s poster “Measuring and controlling dissolution rates of pharmaceutical materials by co-crystal formation” reported dissolution rate studies of tablets of pure paracetamol and co-crystalline forms, and shows a significant increase in dissolution rate of the active ingredient for certain formulations.
James’ poster “Co-crystal or salt? Studying partial proton transfer in a series of molecular materials” reported several crystalline materials which exhibit transfer of a single proton between a significant fraction of the molecular pairs, resulting in a crystal on the borderline between co-crystal and salt.
 The CRYSTALS v14.6236 installer is now available.
The CRYSTALS v14.6236 installer is now available.
This update fixes a number of bugs and eliminates a few mysterious crashes. Please report problems. The minor component or the version number now refers to a specific snapshot of the source code enabling us to better identify and fix bugs.
Key changes between v14.5841 and 14.6236
Ensure copied text is flushed to clipboard on CRYSTALSÂ exit (otherwise it gets lost).
Upgrade xinvert script to accommodate the 11 enantiomorphic space groups and the 7 which require origin shifts.
Diffin: Set default for Z in case no formula found.
Diffin: Extend filename length to 512 to help when running in folders with long paths
Catch crash on matrix inversion of singular matrices.
Xcif: Disable ‘Include HKLI’ and ‘Include squeeze’ if include files not present in folder.
Xafour: Change limiting density question to a dialog to avoid new user confusion.
Close xpublish window after CIF dialog is dismissed.
Add CHANGE RESIDUE to pop-up menus
Speed up plots of internal vs external sigmas
Fix style and centre of rotation of zoomed objects. Add more ‘zoom’ and ‘unzoom’ options to menus
Improve popup text on model toolbar buttons
Add SWAP directive to #EDIT. This swaps the atomic and thermal parameters of pairs of atoms.
Insert residues numbers of generated hydrogen atoms
Sanity check of parameter values when loading from disk. Replace NaN with 0.0 or 1.0 as appropriate.
Show scale factor for electron densities, fix NANs in(Fo^20Fc^2) pattersons.
Cameron: Keep outline of small atoms at smaller scales.
Added header to design matrix ASCII output
Tabbed plot of Internal vs External variances
Insert appropriate occupancy and part # for H’s adjacent to split groups.
Spot clashing H-serial numbers on the fly and correct.
Increase allocation for lexical list processing (was falling over at 500 CONTINUATION lines, should now stretch to 2000).
Extend scattering factor look-up table out to Rho = 3.0
Enable unusual anomalous scattering factors to be input directly from the Cell/SG tab.
Add code to List 12 processing to catch singularities in rigid body code. Least useful rotational parameters are removed from the least-squares.
Enable LIST 2 to be output in full (#PUNCH 2) or just as the symbol (#PUNCH 2 B). This latter enables CRYSTALS to create an appropriate LIST 14 on re-input, otherwise the user must create it.
Changed handling of reflection sigmas. #LIST 6 now has an extra item, L30UP, which controls whether LIST 30 is updated as reflections are read in.
xregulh left SCPQ open, which meant that xwrite5 failed to delete it, resulting in the H optimisation list of refinable atoms being left in there and used to setup L12 if, and only if, there were no OH or NH atoms to be refined. FIXED.
Context menu entry to swap two atom labels.
Read SHELX T .res files (ignore peak height – last column in atom-coordinate list)
Added EXPORT button to FO vs FC graph (csv format).
Extended support for 6-digit H atom serial numbers to xwrite5.scp (not supported everywhere, but here is quite critical).
Improve treatment of A&B parts. Phase shift is included in A&B as they are stored for compatibility with PLATON SQUEEZE, then removed as they are used and the Phase shift applied separately.
Provide clear warning when link for PLATON fails because of a missing LIST
Fix reported cell volume e.s.d for tetragonal and cubic systems (includes correlation of length parameters).
NEW #PUNCH 6 I punches all stored keys.
Use bond list for auto H addition. Use part numbers to generate multiple H conformations on nearby atoms.
Manual ADD H (right-click Add hydrogens) now puts the new H’s into a sensible PART based on connected atom parts. Quite useful.
Allow automatic ‘ride’ of multiple part H’s on same carbon atom (previous version repeated the pivot atom in the constraints list).
Keeping CheckCIF happy: Suppress ESD output on H-bond angle (D-H–A) if the H is riding or fixed.
Add KEYS to reflection statistic plots. Add +/- 10% bands to npp.